37 Genomic ranges and ATAC-seq
Where is the DNA in a cell actually open for business? Most of the genome is wound tightly around nucleosomes and is functionally silent at any given moment. The stretches a cell is actively using — promoters of active genes, enhancers, transcription-factor binding sites — are comparatively accessible. In this chapter you’ll take real sequencing data from an experiment that measures exactly that accessibility, read it into Bioconductor, and walk it through the steps that turn a pile of aligned reads into a biological picture: where reads pile up, how long the sequenced fragments are, and how that fragment length reveals the positions of nucleosomes around the start of every gene.
This chapter builds on the genomic-ranges ideas from the ranges chapter — GRanges, coverage, and the run-length encoding that makes genome-wide signals compact. We’ll lean on those fundamentals rather than re-teach them, and put them to work on a complete ATAC-seq mini-analysis.
R / Bioconductor packages used
- Rsamtools
- GenomicRanges
- GenomicFeatures
- GenomicAlignments
- rtracklayer
- heatmaps
-
TxDb.Hsapiens.UCSC.hg19.knownGene (a large annotation download; install once with
BiocManager::install())
37.1 What you’ll learn
After working through this chapter, you’ll be able to:
- Read a paired-end ATAC-seq BAM file into Bioconductor with
readGAlignmentPairs(), filtering reads as you read. - Summarize reads per chromosome and compute genome-wide coverage as a run-length-encoded signal.
- Examine the fragment-length distribution and explain what nucleosome-free and mononucleosome fragments tell you about chromatin.
- Split reads by fragment length and build TSS heatmaps showing that nucleosome-free reads enrich at transcription start sites.
- Export a coverage track as a BigWig file for viewing in IGV.
37.2 Background
Chromatin accessibility assays measure the extent to which DNA is open and accessible. Such assays now use high throughput sequencing as a quantitative readout. DNase assays — first using microarrays (Crawford, Davis, et al. 2006) and then DNase-seq (Crawford, Holt, et al. 2006) — require more input DNA and are labor-intensive, and have been largely supplanted by ATAC-seq (Buenrostro et al. 2013).
The Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) method maps chromatin accessibility genome-wide. This method quantifies DNA accessibility with a hyperactive Tn5 transposase that cuts and inserts sequencing adapters into regions of chromatin that are accessible. High-throughput sequencing of the fragments produced maps these to regions of increased accessibility, transcription-factor binding sites, and nucleosome positioning. The method is both fast and sensitive and can be used as a replacement for DNase and MNase.
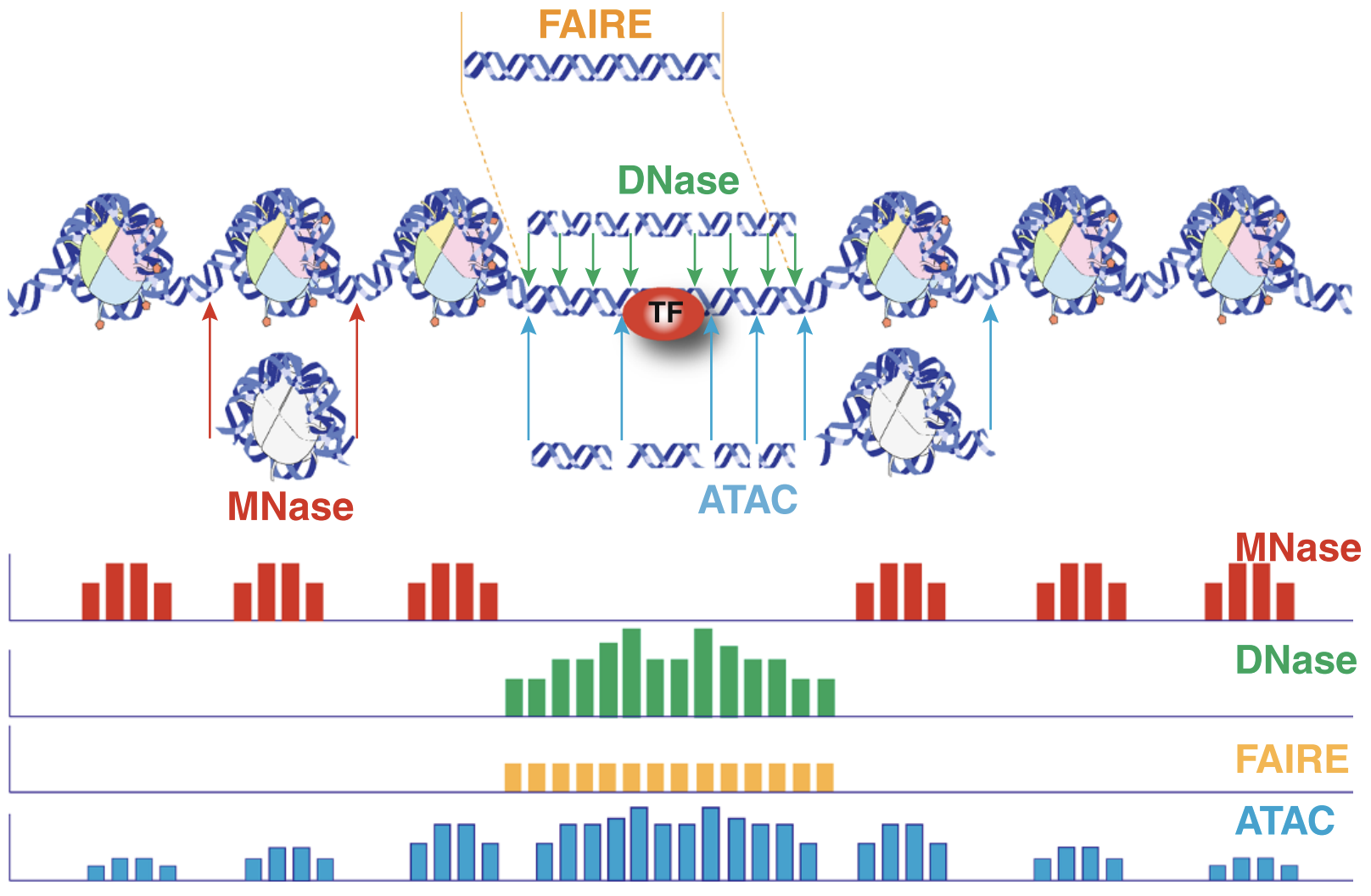
An early review of chromatin accessibility assays (Tsompana and Buck 2014) compares the use cases, pros and cons, and expected signals from each of the most common approaches (Figure 37.1).
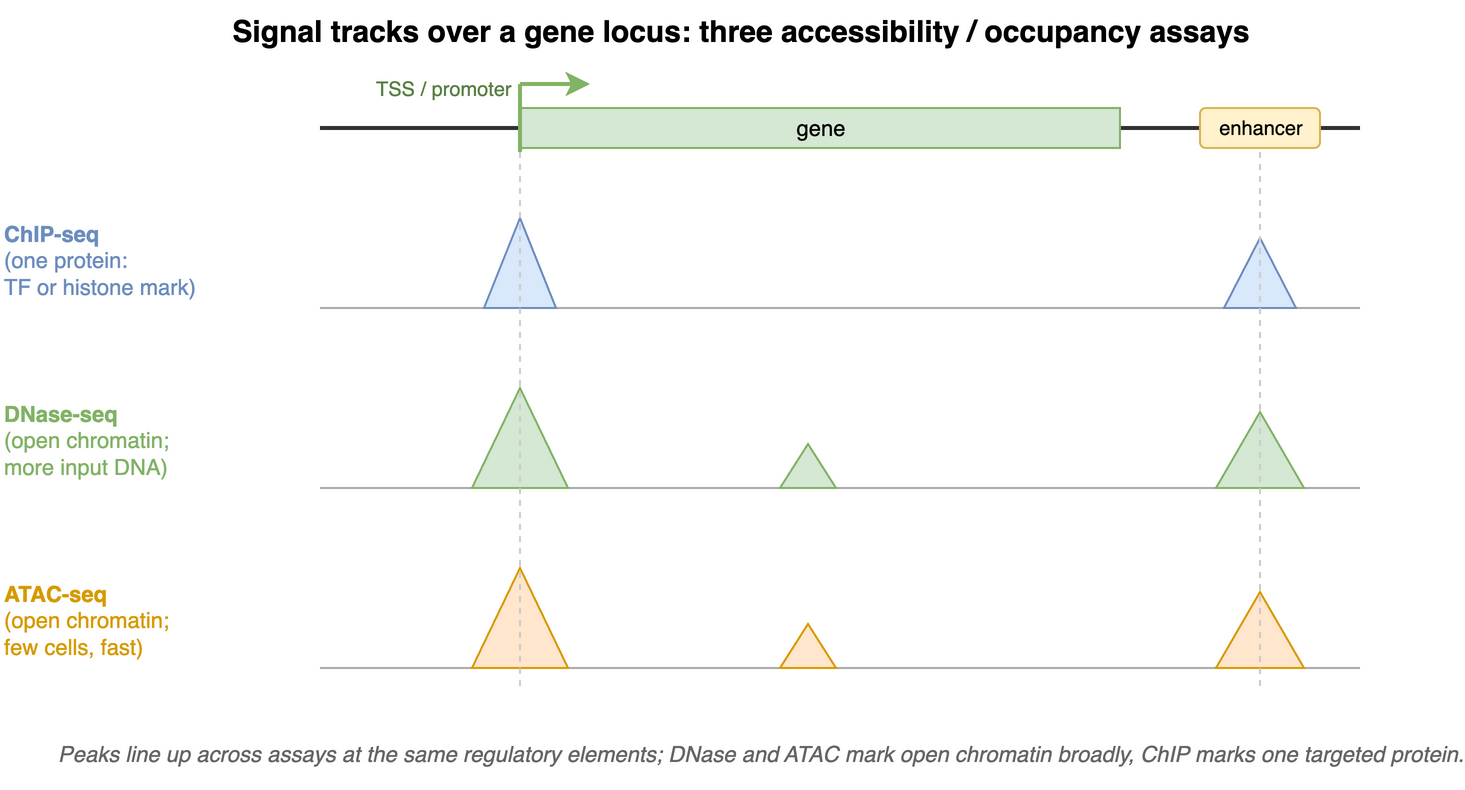
The first manuscript describing the ATAC-seq protocol showed how ATAC-seq data “line up” with other data types such as ChIP-seq and DNase-seq (Figure 37.2). It also highlighted how fragment length correlates with specific genomic regions and characteristics (Buenrostro et al. 2013, fig. 2).
Buenrostro et al. provide a detailed protocol for performing ATAC-Seq and quality control of results (Buenrostro et al. 2015). Updated and modified protocols that improve on signal-to-noise and reduce input DNA requirements have been described.
37.3 Informatics overview
ATAC-seq protocols typically use paired-end sequencing. The reads are aligned to the relevant genome with bowtie2, BWA, or another short-read aligner. After appropriate manipulation — often with samtools — the result is a BAM file (Figure 37.3). Among other details, the BAM format records, for each read:
knitr::include_graphics('imgs/bam_shot.png')
samtools view is the text representation of a BAM file (called SAM format). Bioconductor and many other tools use BAM files for input. BAM files also often have a companion index .bai file that enables random access into the file, so you can read just one genomic region without reading the whole thing.
- sequence name (
chr1) - start position (integer)
- a CIGAR string that describes the alignment in a compact form
- the sequence to which the pair aligns
- the position to which the pair aligns
- a bit flag field that describes multiple characteristics of the alignment
- the sequence and quality string of the read
- additional tags that tend to be aligner-specific
Duplicate fragments (those with the same start and end position of other reads) are marked and likely discarded. Reads that fail to align “properly” are also often excluded from analysis. It is worth noting that most software packages allow simple “marking” of such reads and that there is usually no need to create a special BAM file before proceeding with downstream work.
After alignment and BAM processing, the workflow can switch to Bioconductor.
37.4 Working with sequencing data in Bioconductor
The Bioconductor project includes several infrastructure packages for dealing with ranges (sequence name, start, end, +/- strand) on sequences (Lawrence et al. 2013) as well as capabilities with working with Fastq files directly (Morgan et al. 2016).
| Package | Use cases |
|---|---|
| Rsamtools | low level access to FASTQ, VCF, SAM, BAM, BCF formats |
| GenomicRanges | Container and methods for handling genomic regions |
| GenomicFeatures | Work with transcript databases, gff, gtf and BED formats |
| GenomicAlignments | Reader for BAM format |
| rtracklayer | import and export multiple UCSC file formats including BigWig and Bed |
As noted above, the output of an ATAC-seq experiment is a BAM file. Because paired-end sequencing is the standard for ATAC-seq, readGAlignmentPairs() is the right function to read it.
37.5 Data import and quality control
Reading a paired-end BAM file looks a little involved, but the following code will:
- Read the included BAM file.
- Include read pairs only (
isPaired = TRUE) - Include properly paired reads (
isProperPair = TRUE) - Include reads with mapping quality >= 1
- Add a couple of additional fields,
mapq(mapping quality) andisize(insert size) to the default fields.
greenleaf <- readGAlignmentPairs(
"https://github.com/seandavi/RBiocBook/raw/main/atac-seq/extdata/Sorted_ATAC_21_22.bam",
param = ScanBamParam(
mapqFilter = 1,
flag = scanBamFlag(
isPaired = TRUE,
isProperPair = TRUE
),
what = c("mapq", "isize")
)
)The object we just created, greenleaf, holds every read pair that passed our filters, with the start and end of each fragment and the two extra columns we asked for. (The Exercises at the end of the chapter ask you to inspect it more closely.)
Now let’s get a first sense of the data. A simple, useful quality-control check is the number of reads mapping to each chromosome.
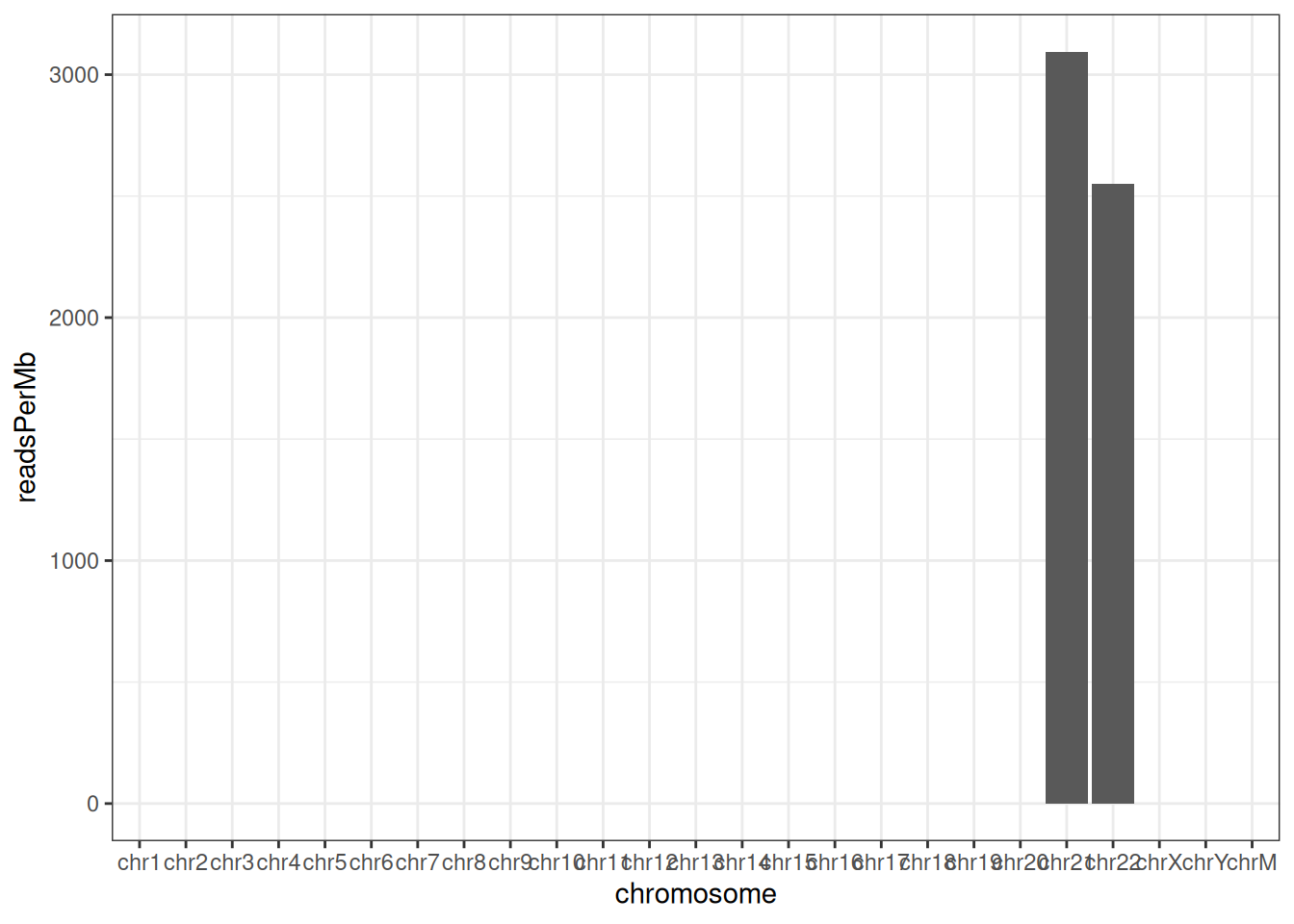
To keep the example small, this BAM file includes only chromosomes 21 and 22, so Figure 37.4 shows just those two bars. With a full genome you’d see one bar per chromosome, and a chromosome with far fewer (or far more) reads than its neighbors can be an early sign of a problem.
ggplot(chromCounts, aes(x = chromosome, y = count)) +
geom_bar(stat = "identity") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))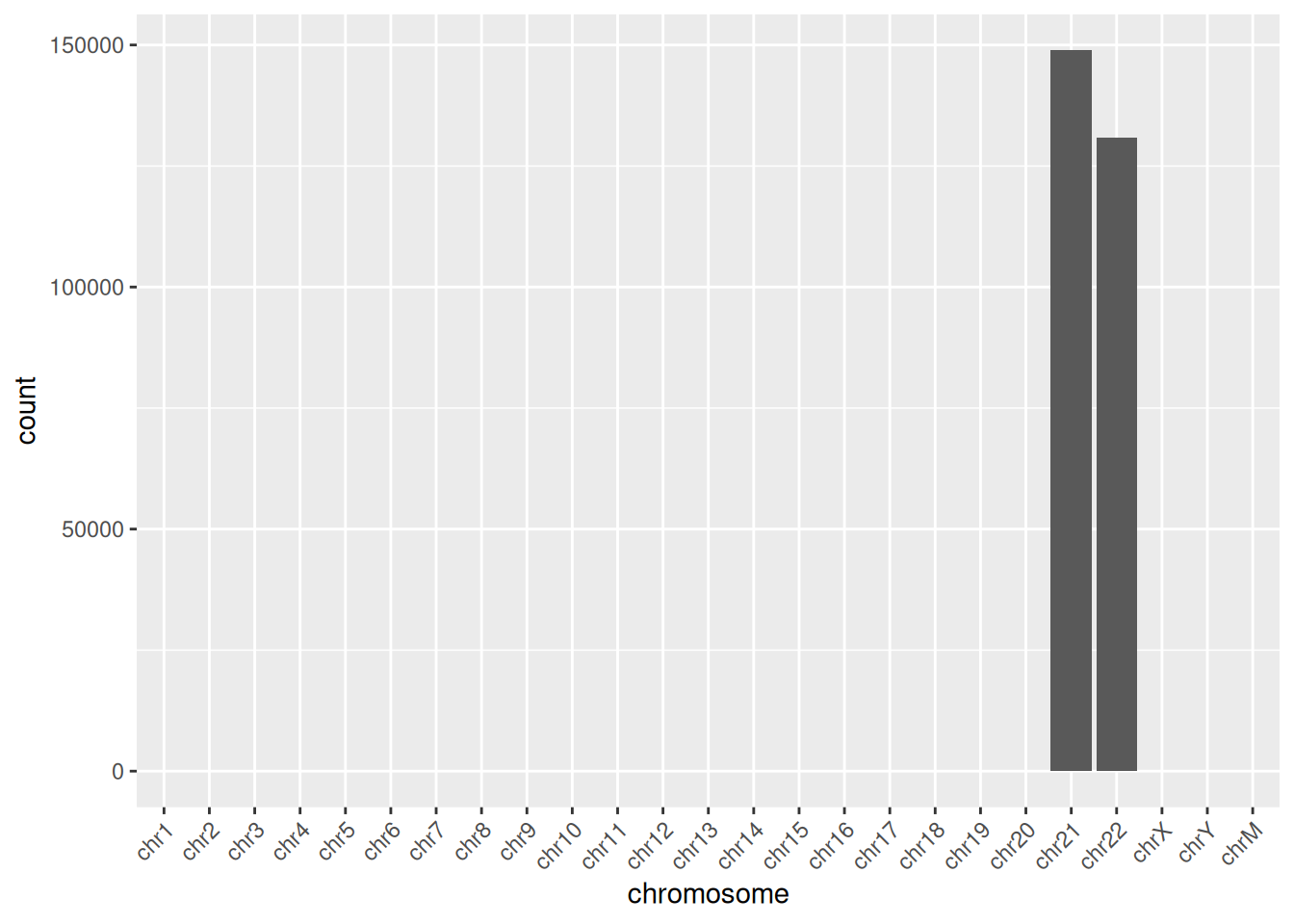
Raw counts are hard to compare across chromosomes because longer chromosomes naturally collect more reads. Normalizing by chromosome length gives reads per megabase, which should be roughly similar across chromosomes if coverage is even.
chromCounts <- chromCounts %>%
dplyr::mutate(readsPerMb = (count / (seqlengths(greenleaf) / 1e6)))With only two chromosomes the normalized plot (Figure 37.5) is a little underwhelming, but the same idea scales to a whole genome.
37.6 Coverage
The coverage() method for genomic ranges counts, at each base, how many features overlap that position. For our ATAC-seq read pairs, the coverage tells us how reads pile up — and tall piles are the peaks that mark accessible chromatin.
Coverage comes back as a SimpleRleList. The names() function gives us the names of its elements.
names(cvg) [1] "chr1" "chr2" "chr3" "chr4" "chr5" "chr6" "chr7" "chr8" "chr9"
[10] "chr10" "chr11" "chr12" "chr13" "chr14" "chr15" "chr16" "chr17" "chr18"
[19] "chr19" "chr20" "chr21" "chr22" "chrX" "chrY" "chrM" There is one element per chromosome. Let’s look at the chr21 entry:
cvg$chr21integer-Rle of length 48129895 with 397462 runs
Lengths: 9411376 50 11 50 ... 36 14 28 10806
Values : 0 2 0 2 ... 1 2 1 0Rle?
Each chromosome’s coverage is stored as an Rle, short for run-length encoding. Along a chromosome the coverage value stays the same for long stretches — millions of bases in a row might all be 0. Instead of storing each base separately, an Rle records (run length : value) pairs. If the first 9,410,000 bases all have coverage 0, that’s stored as a single pair (9,410,000 : 0) rather than nine-million-plus zeros. Across a whole genome this saves an enormous amount of memory, which is why genome-wide signals in Bioconductor are almost always run-length encoded.
The little function plotCvgHistByChrom() below plots a histogram of the coverage values seen on a chromosome — how many bases have coverage 0, coverage 1, coverage 2, and so on.
library(ggplot2)
plotCvgHistByChrom <- function(cvg, chromosome) {
cvgcounts <- as.data.frame(table(cvg[[chromosome]]))
cvgcounts[, 1] <- as.numeric(as.character(cvgcounts[, 1]))
colnames(cvgcounts) <- c("Coverage", "Count")
ggplot(cvgcounts, aes(x = Coverage, y = Count)) +
ggtitle(paste("Chromosome", chromosome)) +
geom_point(alpha = 0.5) +
geom_smooth(span = 0.2) +
scale_y_log10() +
theme_bw()
}Drawing it for both chromosomes (Figure 37.6):
plotCvgHistByChrom(cvg, "chr21")
plotCvgHistByChrom(cvg, "chr22")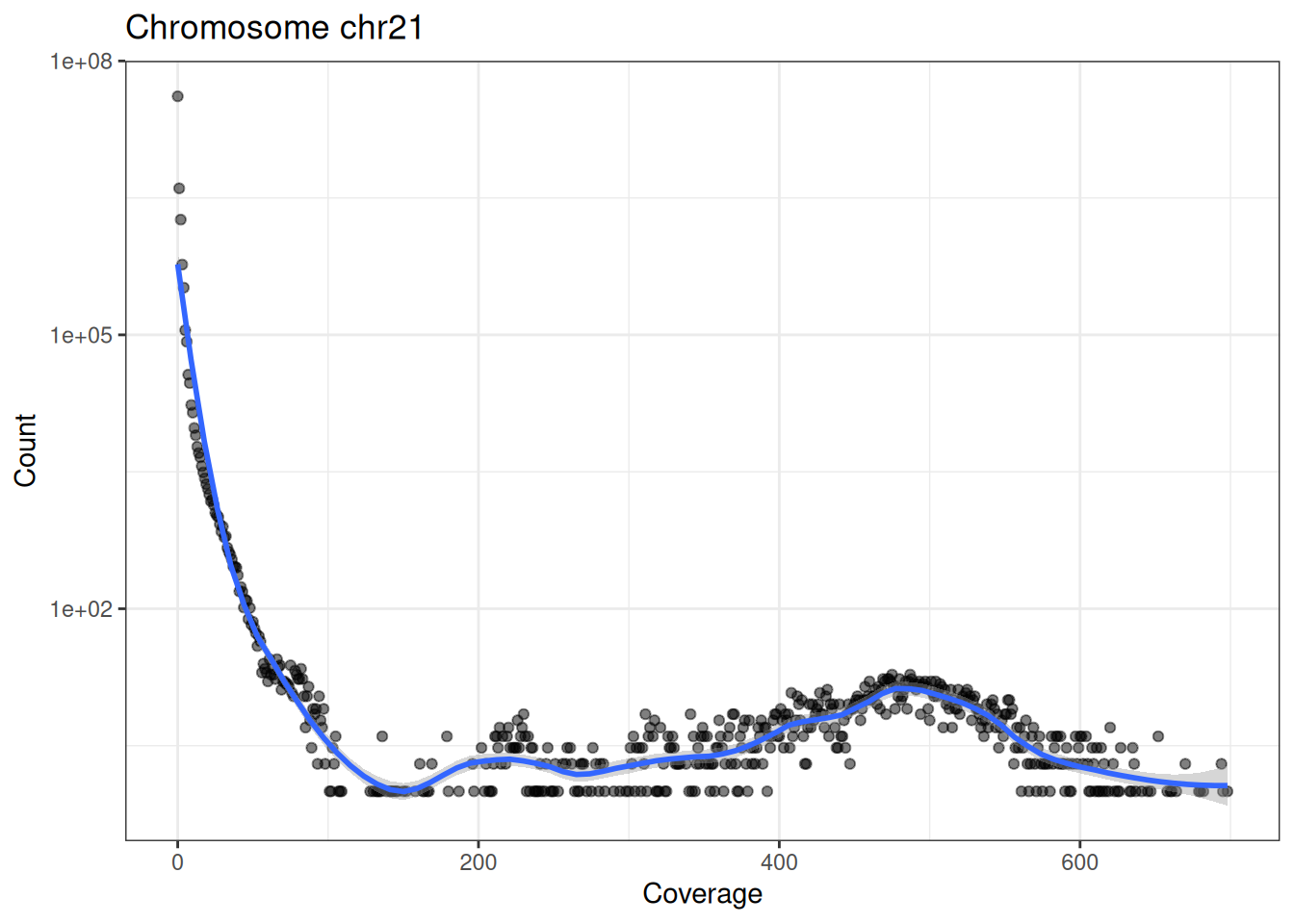
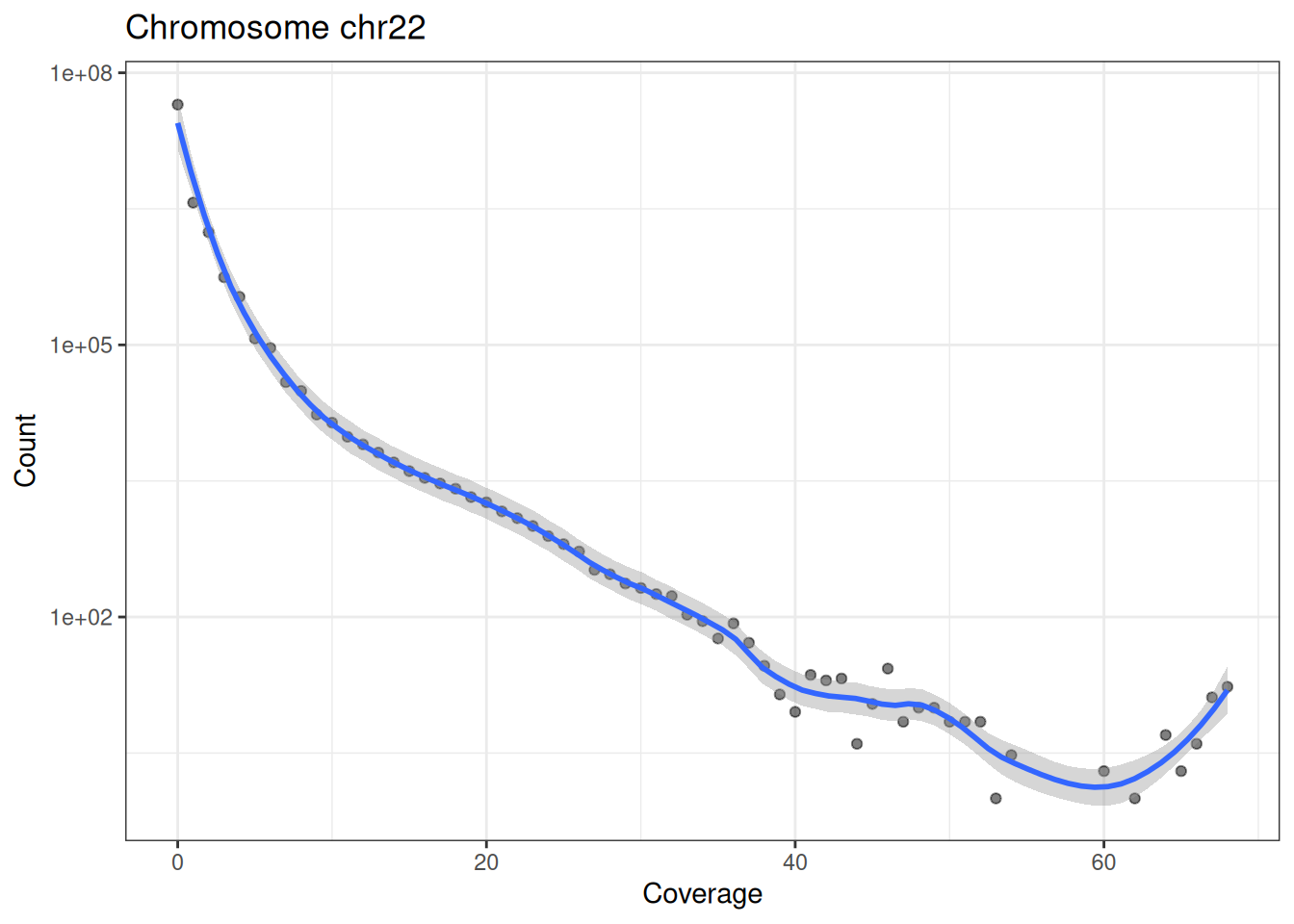
In Figure 37.6 (note the log-scaled y axis) the vast majority of bases have very low coverage, and the count drops sharply as coverage rises. That long right-hand tail of high-coverage bases is exactly what we care about — those are the accessible regions where reads accumulate into peaks.
37.7 Fragment lengths
The first ATAC-seq manuscript (Buenrostro et al. 2013) highlighted a beautiful relationship between fragment length and nucleosomes (Figure 37.7).
knitr::include_graphics("imgs/atac_fragment_length.png")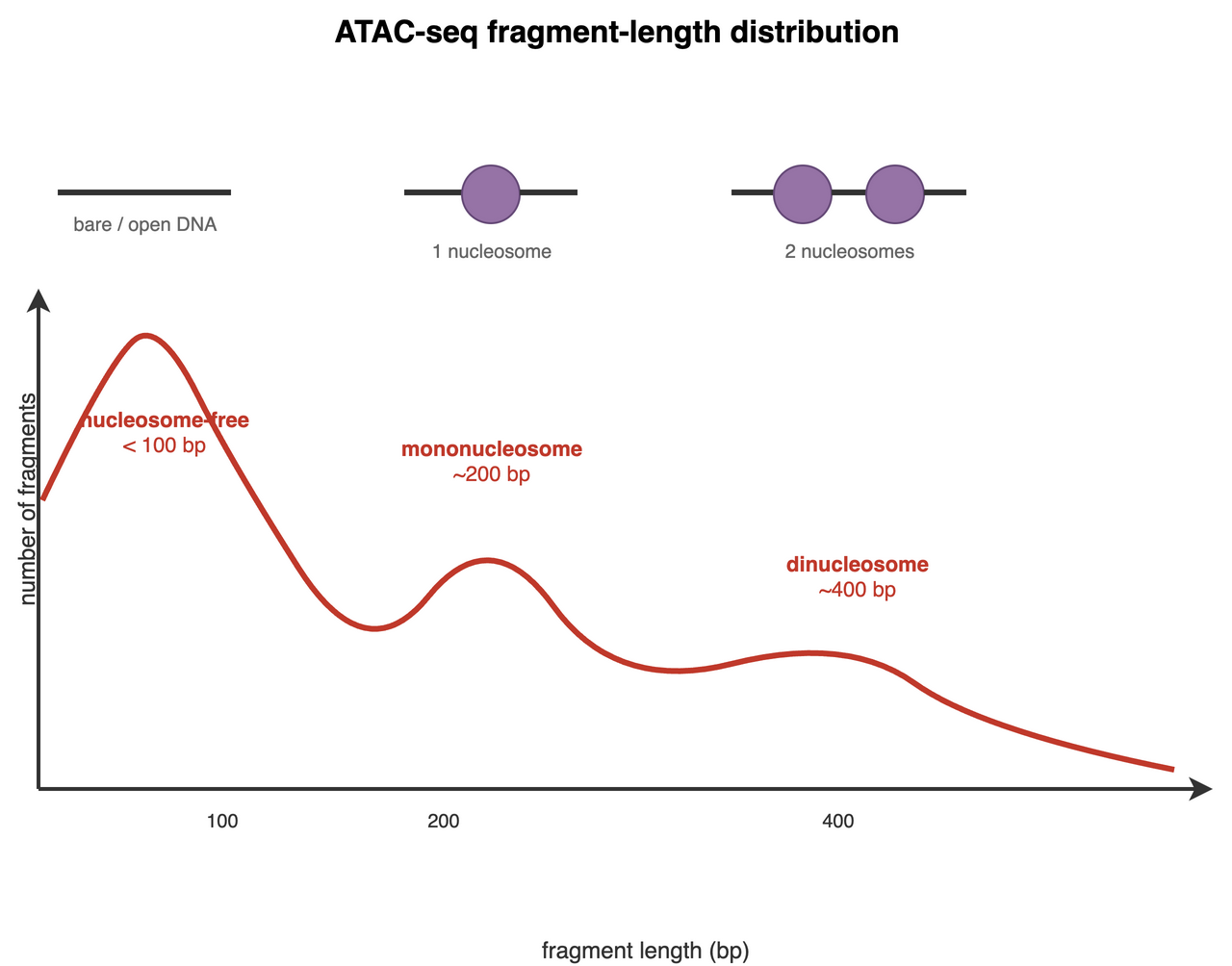
The Tn5 transposase can only cut DNA where it can physically reach it — between nucleosomes, not through them. So the distance between two cut sites (the fragment length, also called insert size) tells you how much chromatin sat between them:
- Nucleosome-free fragments are short, shorter than ~147 bp (one nucleosome’s worth of DNA). These come from open regions — promoters, enhancers, accessible regulatory DNA.
- Mononucleosome fragments are roughly 147–294 bp: long enough to have wrapped one nucleosome.
- Longer fragments span two or more nucleosomes, giving the periodic ~147 bp ladder you see in Figure 37.7.
Because active promoters are nucleosome-free right at the transcription start site (TSS), the short, nucleosome-free reads should pile up at the TSS, while mononucleosome reads should flank it. We’ll confirm exactly that with a heatmap at the end of the chapter.
We loaded the BAM file with insert sizes (isize), so we can read fragment lengths straight off that column. The insert size is the same (in absolute value) for the first and second read of a pair, so we’ll just use first.
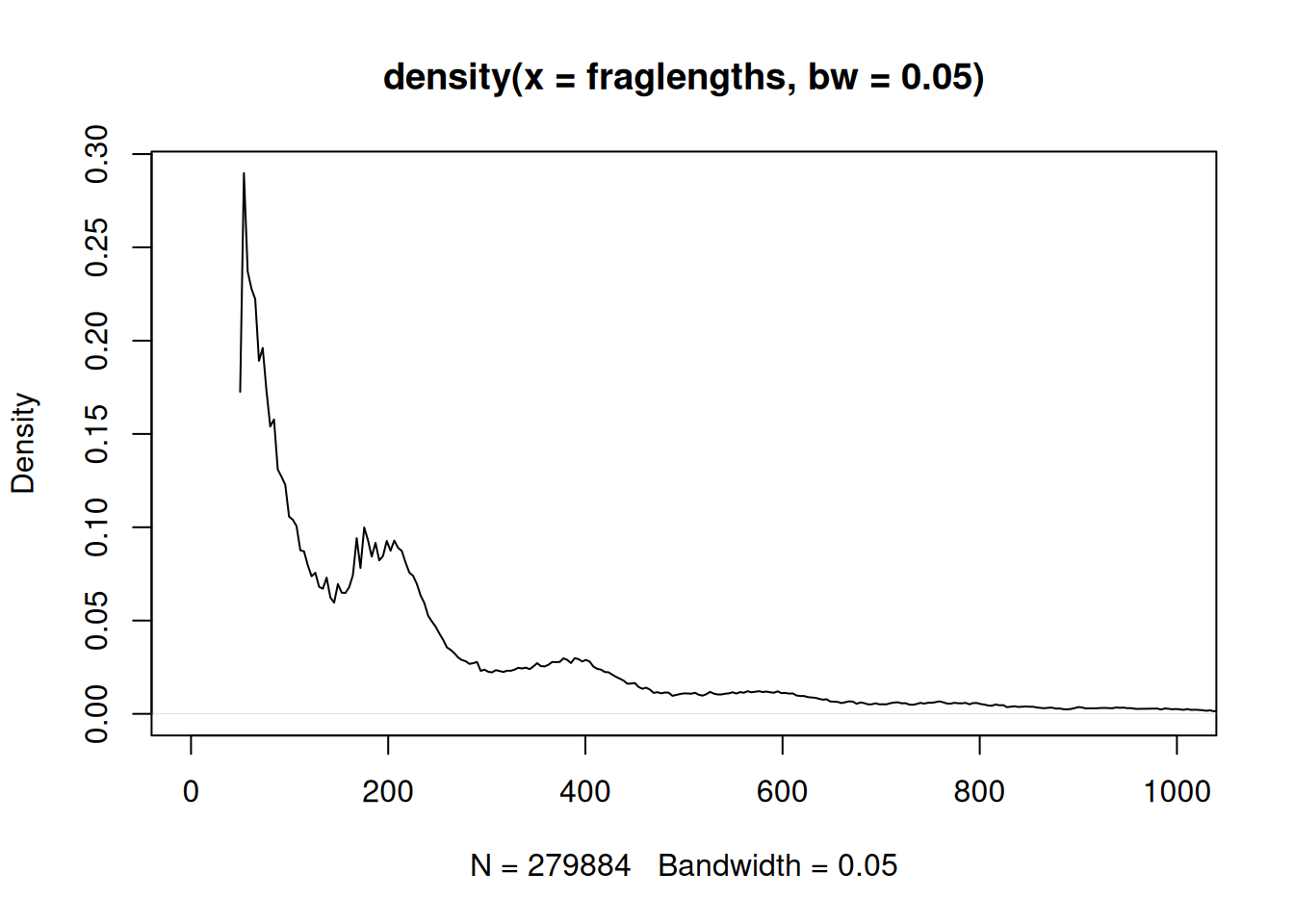
Now we can look at the distribution of fragment lengths. The density() function gives a smoothed version (Figure 37.8).
A ggplot2 version of the same distribution (Figure 37.9) makes the periodic structure easier to read:
library(dplyr)
library(ggplot2)
fragLenPlot <- table(fraglengths) %>%
data.frame() |>
rename(
InsertSize = fraglengths,
Count = Freq
) |>
mutate(
InsertSize = as.numeric(as.vector(InsertSize)),
Count = as.numeric(as.vector(Count))
) |>
ggplot(aes(x = InsertSize, y = Count)) +
geom_line()
print(fragLenPlot + theme_bw() + lims(x = c(-1, 500)))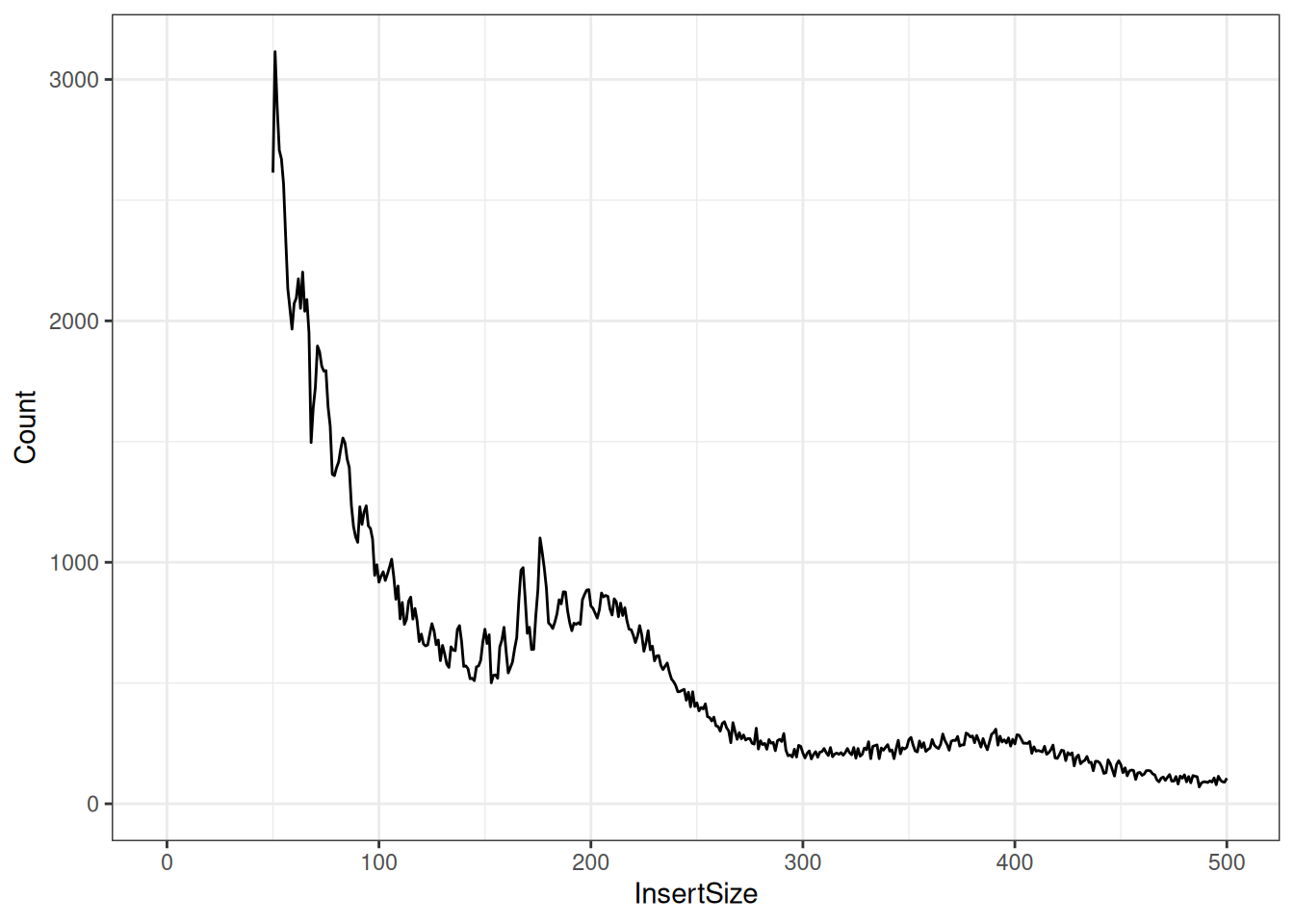
Look at the shape: a tall peak at short fragment lengths, then a smaller hump around 200 bp. That first peak is the nucleosome-free DNA, and the hump is the mononucleosome fragments described in the callout above. This bimodal shape is one of the standard quality checks for an ATAC-seq library — a healthy library shows it clearly.
Because we can read each fragment’s length, we can split the reads by their biology. First, the nucleosome-free reads, with insert size shorter than one nucleosome:
And the mononucleosome reads, with insert size between 147 and 294 bp:
Now for the payoff. If the biology in the callout holds, the nucleosome-free reads should be enriched right at the TSS, while the mononucleosome reads should not be. We’ll use the heatmaps package to compare these two sets of reads against the transcription start sites of the human genome.
Here proms is a set of windows reaching 250 bp on each side of every annotated TSS on chromosomes 21 and 22 — one row per promoter for the heatmaps to come. The heatmaps package vignette describes the package’s full capabilities if you’d like to go further.
We build one heatmap per read set, lining up the coverage over those promoter windows and scaling both to the same range so they’re directly comparable.
The “meta” plot in Figure 37.10 averages the signal across all promoters, giving one summary curve per read set.
gl_mn_hm <- CoverageHeatmap(proms, coverage(gl_mn), coords = c(-250, 250))
label(gl_mn_hm) <- "MonoNuc"
scale(gl_mn_hm) <- c(0, 10)
plotHeatmapMeta(list(gl_nf_hm, gl_mn_hm))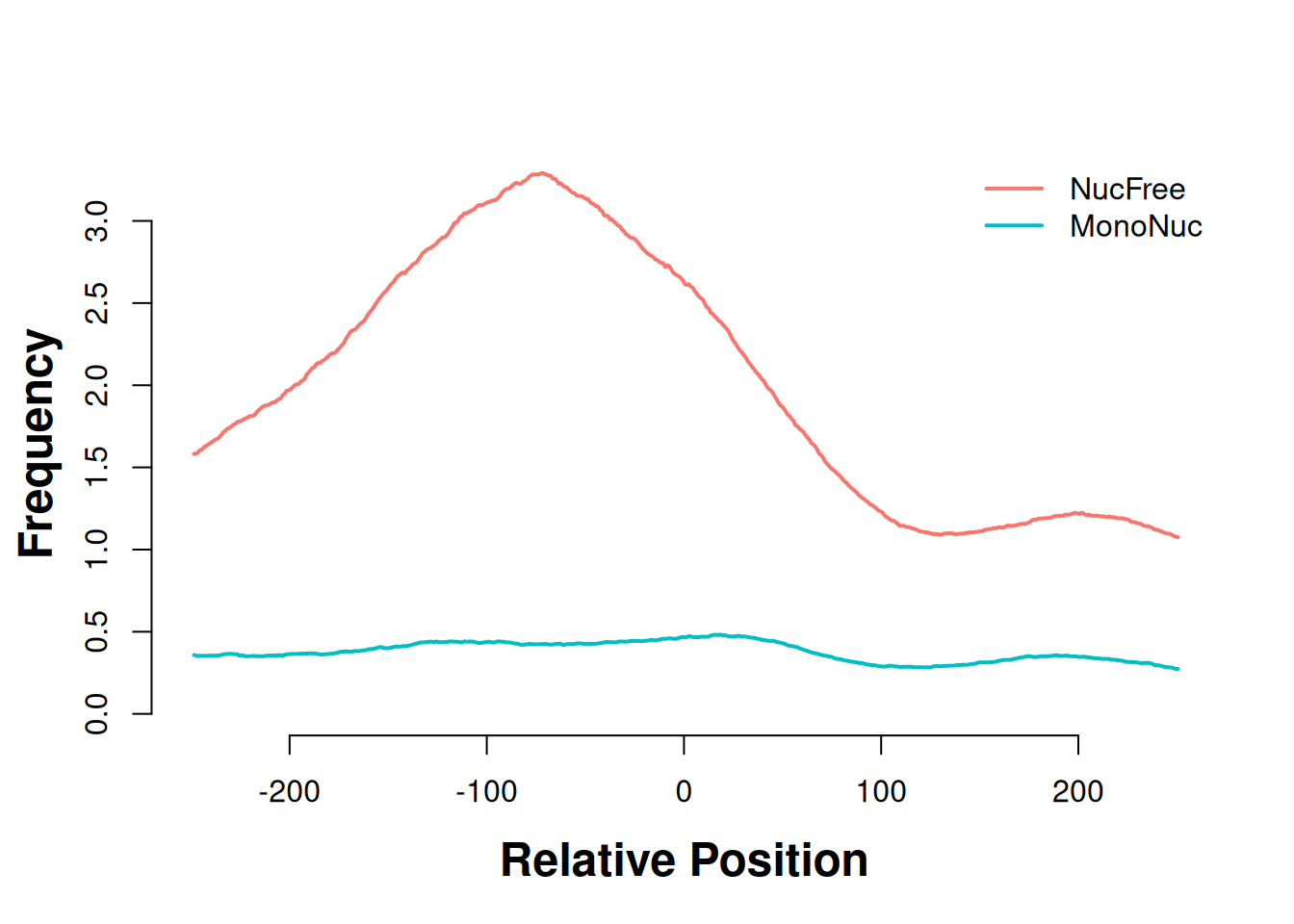
This is the result the callout predicted: the nucleosome-free curve spikes right at the TSS (position 0), while the mononucleosome curve barely moves. The full heatmaps in Figure 37.11 show the same thing promoter-by-promoter — each row is one TSS, and the bright vertical band in the nucleosome-free panel is the accessible signal stacking up at the start of gene after gene.
plotHeatmapList(list(gl_mn_hm, gl_nf_hm))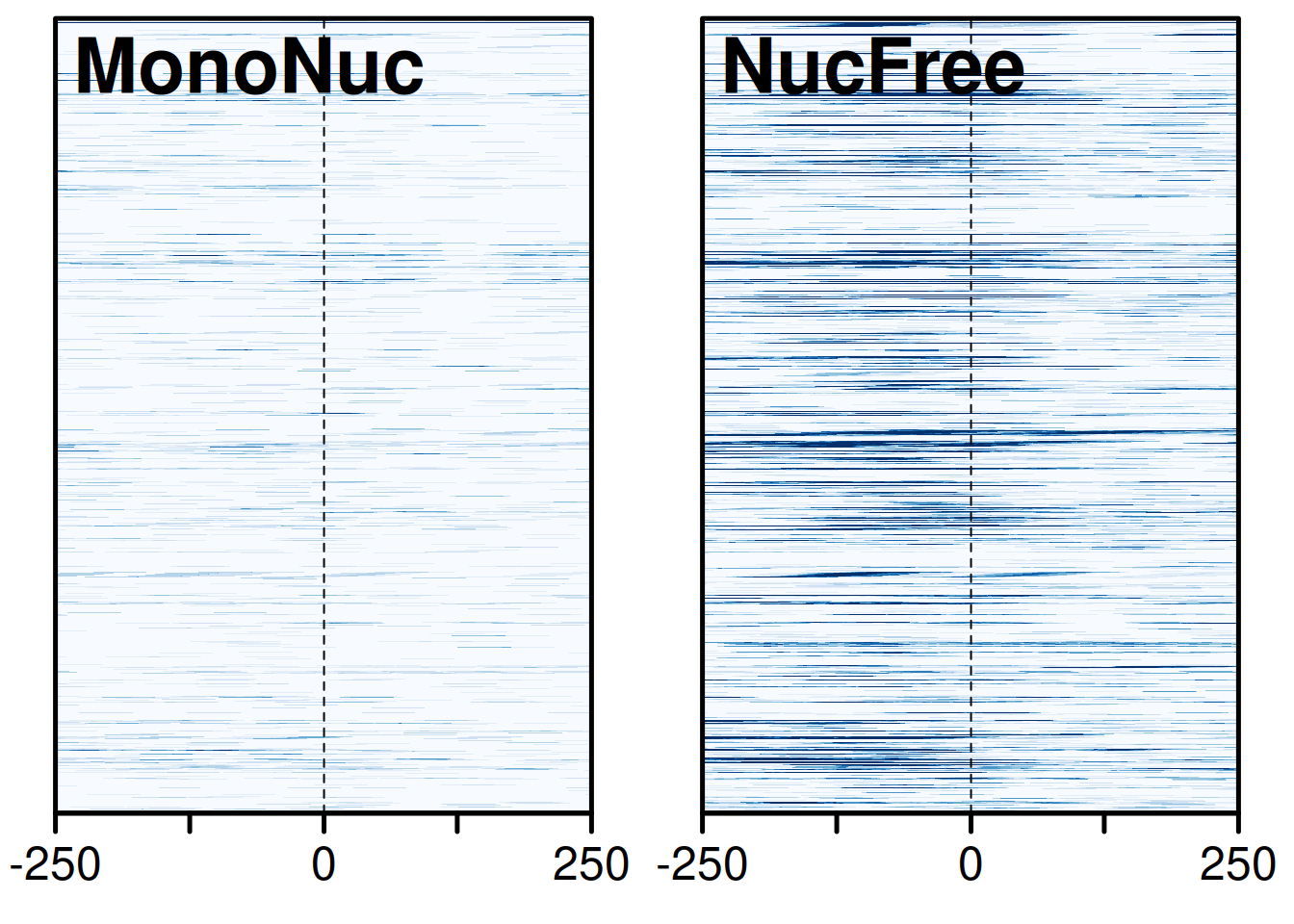
37.8 Viewing data in IGV
A heatmap summarizes thousands of regions at once, but sometimes you want to scroll along the genome and look at the pileups. The Integrative Genomics Viewer (IGV) is the standard tool for that. Install it from the Broad Institute.
To load our data into IGV, we export the coverage as a BigWig file — a compact, indexed format IGV reads quickly.
The Exercises below ask you to open this track in IGV and explore it.
37.9 Exercises
These exercises build on the greenleaf object you read in above. The first three are short coding tasks with solutions; the last two are hands-on exploration in IGV.
-
What kind of object is
greenleaf? Find its class.NoteSolutionclass(greenleaf)[1] "GAlignmentPairs" attr(,"package") [1] "GenomicAlignments"readGAlignmentPairs()returns aGAlignmentPairsobject — a container that keeps the two mates of each read pair together. -
Get the first read of each pair. Use the
GenomicAlignments::first()accessor to pull the first read of every pair as aGAlignmentsobject, save it asgl_first_read, then usemcols()to look at its metadata columns.NoteSolutiongl_first_read <- GenomicAlignments::first(greenleaf) gl_first_readGAlignments object with 279884 alignments and 2 metadata columns: seqnames strand cigar qwidth start end width <Rle> <Rle> <character> <integer> <integer> <integer> <integer> [1] chr21 + 50M 50 9411377 9411426 50 [2] chr21 + 50M 50 9411377 9411426 50 [3] chr21 - 50M 50 9411639 9411688 50 [4] chr21 - 50M 50 9413594 9413643 50 [5] chr21 - 50M 50 9413594 9413643 50 ... ... ... ... ... ... ... ... [279880] chr22 + 50M 50 51238222 51238271 50 [279881] chr22 + 50M 50 51238594 51238643 50 [279882] chr22 - 50M 50 51239601 51239650 50 [279883] chr22 + 50M 50 51240499 51240548 50 [279884] chr22 + 50M 50 51240499 51240548 50 njunc | mapq isize <integer> | <integer> <integer> [1] 0 | 40 111 [2] 0 | 40 111 [3] 0 | 20 -123 [4] 0 | 20 -168 [5] 0 | 20 -168 ... ... . ... ... [279880] 0 | 40 205 [279881] 0 | 13 373 [279882] 0 | 20 -98 [279883] 0 | 13 424 [279884] 0 | 10 424 ------- seqinfo: 25 sequences from an unspecified genomemcols(gl_first_read)DataFrame with 279884 rows and 2 columns mapq isize <integer> <integer> 1 40 111 2 40 111 3 20 -123 4 20 -168 5 20 -168 ... ... ... 279880 40 205 279881 13 373 279882 20 -98 279883 13 424 279884 10 424The metadata columns hold the extra fields we requested when reading the BAM —
mapq(mapping quality) andisize(insert size, i.e. fragment length). -
How many read pairs map to each chromosome? Summarize the counts per chromosome.
NoteSolutionchr1 chr2 chr3 chr4 chr5 chr6 chr7 chr8 chr9 chr10 chr11 0 0 0 0 0 0 0 0 0 0 0 chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chr20 chr21 chr22 0 0 0 0 0 0 0 0 0 148940 130944 chrX chrY chrM 0 0 0This is the same count we plotted in Figure 37.4. Only chromosomes 21 and 22 are present in this trimmed example file.
Explore the full track in IGV. Open IGV, choose the
hg19genome, then load thegreenleaf.bwfile you exported. Navigate to chromosomes 21 and 22 and look for the peaks. Do the tallest pileups fall near gene starts?Compare the nucleosome-free track in IGV. Export the nucleosome-free reads (
gl_nf) as their own BigWig file withexport.bw(), load it alongside the full track, and compare. Where do you expect the strongest signals — and does what you see in IGV match the TSS heatmaps from Figure 37.11?
37.10 Summary
You took a paired-end ATAC-seq BAM file all the way from raw alignments to a biological result. You read and filtered the reads with readGAlignmentPairs(), checked reads per chromosome, and computed genome-wide coverage stored compactly as run-length-encoded Rle signals. You then used the fragment-length distribution to separate nucleosome-free from mononucleosome reads, and built TSS heatmaps showing that the nucleosome-free reads enrich sharply at transcription start sites — exactly what the biology of open chromatin predicts. Finally, you exported a coverage track for browsing in IGV. The same workflow scales from these two chromosomes to a whole genome.
37.11 Additional work
If you work extensively with ATAC-seq, Thomas Carroll’s workshop is an excellent next step:
Feel free to work through it. The ATACseqQC package vignette also offers a great deal beyond quality control, and several more ATAC-seq packages are available in Bioconductor.
Calling peaks on the coverage itself — finding the significant pileups rather than just plotting them — is most often done outside R with MACS (pip install macs2).