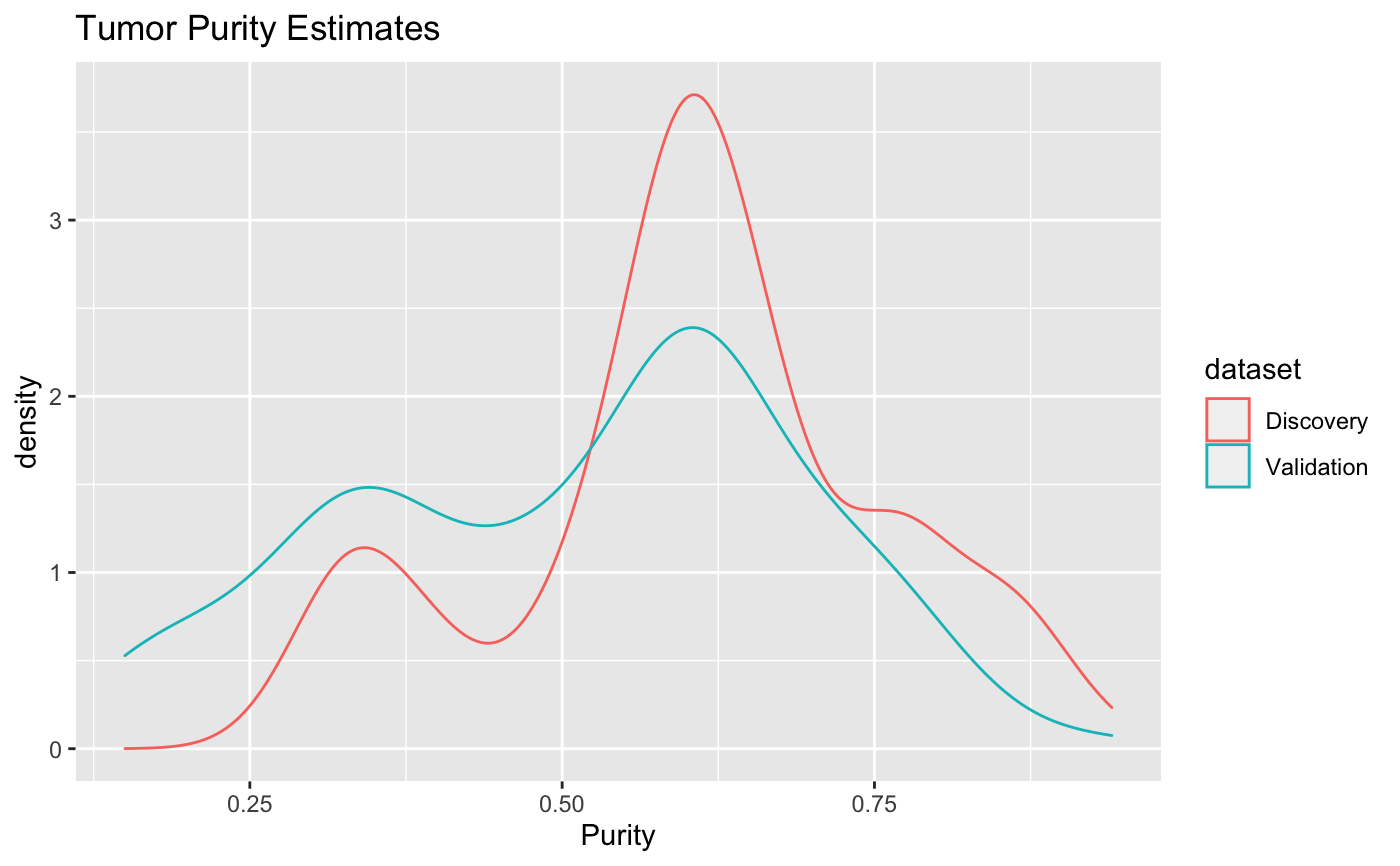
Results from PureCN run on discovery and validation sets.
target_purity()
Value
A data.frame with columns Source,
Purity, Ploidy, and dataset
Examples
pure = target_purity()#> Warning: Missing column names filled in: 'X23' [23], 'X25' [25], 'X26' [26], 'X27' [27], 'X28' [28]#> Warning: Duplicated column names deduplicated: 'SampleName' => 'SampleName_1' [21], 'SampleName' => 'SampleName_2' [22], 'Sample_Name_Roche' => 'Sample_Name_Roche_1' [24]head(pure)#> # A tibble: 6 x 4 #> Source Purity Ploidy dataset #> <chr> <dbl> <dbl> <chr> #> 1 0A4HLD 0.65 2.80 Discovery #> 2 0A4HMC 0.33 2.66 Discovery #> 3 0A4HX8 0.87 5.26 Discovery #> 4 0A4I6O 0.53 1.59 Discovery #> 5 0A4HXS 0.39 2.14 Discovery #> 6 0A4HY5 0.66 2.51 Discovery#> #>#> #> #>#> #> #>#> #> #>#> #> #>#> #> #>#> #> #>#> #> #>#> #> #>#> #> #>#> #> #>pure %>% group_by(dataset) %>% summarise(medploidy = median(Ploidy, na.rm=TRUE), medpurity = median(Purity, na.rm=TRUE), lowpurity = quantile(Purity, 0.1, na.rm=TRUE))#> # A tibble: 2 x 4 #> dataset medploidy medpurity lowpurity #> <chr> <dbl> <dbl> <dbl> #> 1 Discovery 3.10 0.6 0.359 #> 2 Validation 2.92 0.55 0.252library(ggplot2) p = ggplot(pure, aes(x=Purity)) + geom_density(aes(group=dataset, color=dataset)) + ggtitle('Tumor Purity Estimates') print(p)